
ManufacturingApril 25, 2024 · Armelle Chavanet
Dassault Systèmes and HD Hyundai Heavy Industry sign MoU for Virtual Twin-based Integrated Design-Production Platform
Design & SimulationApril 18, 2024 · Taherah KUHL
Virtual Twins: Not just for manufacturing anymore…


Latest articles
Discover our most recent posts across all industries, brands and topics.

The opportunity advantage of early AI adoption
Failure to adopt AI quickly and effectively can present a significant challenge, particularly in consumer-facing industries like manufacturing.
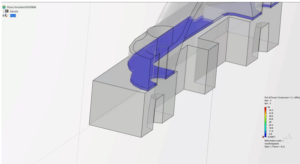
Gasket Simulation in Abaqus
This blog discusses gasket simulation in Abaqus/Standard, focusing on real-world applications. Gaskets provide cost-effective seals between mating surfaces and have applications across various industries.

Meet the mindful mentor who is changing how we think about work
Striving for a strong work life balance and happiness is what inspires Els Van Langenhove to mentor others.

How can we make Earth our most important stakeholder?
This Earth Day, a rapidly advancing combination of science and technology is beginning to let us view Earth as a stakeholder.

What Is The Sales Academy?
For the third year in a row now, we are looking for new candidates for our trainee program, the Sales Academy. But what is behind this program and who is it for? Let’s take a look together.

The secret behind smart home product development: Simplify the complexity
Consumers shopping for home products notice the common home products shifting to “smart” products. It has taken some time, but everyday consumer products now use technology from the aerospace and industrial sectors.

How might a drone save lives?
The Technical University of Munich (TUM) in Germany is one of many universities benefitting from the 3DEXPERIENCE platform so that it can bring industry best-practices into the learning environment.

How to Get Started with MBSE?
Your most common question answered with this practical guide, where we learn and decode the complexities of implementing MBSE.

The secret behind smart home product development: platform-based MBSE
Consumers shopping for home products notice the common home products shifting to “smart” products. It has taken some time, but everyday consumer products now use technology from the aerospace and industrial sectors.

Celebrating Earth Day 2024
The primary goal of Earth Day is to raise awareness about the importance of preserving our planet’s natural resources and promoting sustainability. It’s a day for individuals, communities, and organizations worldwide to participate in activities and initiatives that contribute to environmental conservation.

Navigating Complexities of Radar Vehicle Integration Leveraging Electromagnetic Simulation
Please welcome guest blogger, Yadhukrishnan MK, of Continental AG as he talks about radar simulation in advanced driver assistance systems.
Explore our topics
Discover stories on key trends that are transforming the business and social landscape.
Company News
Keep up with the latest happenings at Dassault Systèmes.
The opportunity advantage of early AI adoption
Failure to adopt AI quickly and effectively can…
How can we make Earth our most important…
This Earth Day, a rapidly advancing combination of…
How might a drone save lives?
The Technical University of Munich (TUM) in Germany…

[Press Release] Dixon Technologies India Signs MoU with Dassault…
This partnership aims to revolutionize the way products…

The ultimate guide to cloud digital transformation in…
Discover how digital transformation using cloud computing and…
Design & Simulation
Today’s challenges are being met with innovative design and manufacturing and improve through simulation.
Gasket Simulation in Abaqus
This blog discusses gasket simulation in Abaqus/Standard, focusing…
How to Get Started with MBSE?
Your most common question answered with this practical…
The secret behind smart home product development: platform-based…
Consumers shopping for home products notice the common…
Navigating Complexities of Radar Vehicle Integration Leveraging Electromagnetic…
Please welcome guest blogger, Yadhukrishnan MK, of Continental…

Virtual Twins: Not just for manufacturing anymore…
Explore how financial services organizations can use Virtual…
Editor’s picks
Explore our favorite stories

Manufacturing
It’s Fall Festival Time! Is Your Supply Chain…
Many event planners and teams focused on the supply chain may also wonder about possible…

Manufacturing
Decoding with Artificial Intelligence and Augmented Reality
New technologies offer considerable opportunities and advantages to industrial players seeking to adapt to the…

Company News
How green is your car?
The way a car’s environmental performance is measured is changing. One German engineering services provider…

Company News
How one company is leading a revolution in…
With a novel approach to reconstructive surgery, LATTICE MEDICAL is helping to shape the next…

Manufacturing
Virtual Twins for Industrial Robotics
While 34% percent of today’s biggest companies have committed to zero-emissions goals, 93% will miss…
Company News
What will healthy mean in 2040?
A new vision emerges for the convergence of life sciences innovation with global healthcare, into…
Design & Simulation
Ask an Engineer: What is Machine Learning and…
An interview with SIMULIA’s Jing Bi, a Technology Senior manager who specializes in physics-based simulation…
Latest articles from brands
Learn more about our portfolio of 3D modeling applications, simulation applications creating virtual twins, social and collaborative applications, and information intelligence applications.

The Dassault Systémes Marketing and Sales brand 3DEXCITE provides comprehensive solutions for collaboration and 3D product content creation directly from a secure cloud network, the 3DEXPERIENCE® platform.

BIOVIA is the world’s leading solution for product design and experience. It delivers the unique ability not only to model any product in 3D but to do so in the context of the products real life behavior.

CATIA provides product design and experience capabilities for designers, engineers and system architects to conceive, develop and realize the connected products and experiences we see and use in our everyday lives.
Design & Simulation
How to Get Started with MBSE?

Powered by Dassault Systèmes’ 3DEXPERIENCE platform, DELMIA solutions address the most challenging situations manufacturers experience today. We connect the virtual and real worlds with immersive technology to empower our customers worldwide to collaborate, model, optimize, and execute manufacturing, logistics, and service across an efficient and connected supply chain.
Manufacturing
Celebrating Earth Day 2024

ENOVIA provides enterprise planning, IP management, process governance, and collaborative product development capabilities for multi-disciplinary teams to collaboratively build and execute a successful plan across the entire enterprise.


GEOVIA provides modelling, designing, simulation and monitoring for geoscientists, earth engineers, urban planners and citizens to enable the sustainable use and reuse of natural resources across urban environments and the broader infrastructure sector.

Sustainability
From Mining Data to Mining Intelligence

NETVIBES provides data science capabilities that reveal information intelligence for all business users to gain business insights that drive performance.


SIMULIA provides realistic multiphysics simulation, design exploration, and optimization capabilities for designers, engineers and researchers to virtually test, improve and validate their innovations for performance, safety and consumer experiences while reducing cost and time to market.
Design & Simulation
Gasket Simulation in Abaqus
Stay up to date